Continued from previous page
The atmospheric abundance of CH4 has increased by about a factor
of 2.5 since the pre-industrial era (see Figure 4.1a)
as evidenced by measurements of CH4 in air extracted from ice cores
and firn (Etheridge et al., 1998). This increase still continues, albeit at
a declining rate (see Figure 4.1b). The global tropospheric
CH4 growth rate averaged over the period 1992 through 1998 is about
4.9 ppb/yr, corresponding to an average annual increase in atmospheric burden
of 14 Tg. Superimposed on this long-term decline in growth rate are interannual
variations in the trend (Figure 4.1c). There are
no clear quantitative explanations for this variability, but understanding these
variations in trend will ultimately help constrain specific budget terms. After
the eruption of Mt. Pinatubo, a large positive anomaly in growth rate was observed
at tropical latitudes. It has been attributed to short-term decreases in solar
UV in the tropics immediately following the eruption that decreased OH formation
rates in the troposphere (Dlugokencky et al., 1996). A large decrease in growth
was observed, particularly in high northern latitudes, in 1992. This feature
has been attributed in part to decreased northern wetland emission rates resulting
from anomalously low surface temperatures (Hogan and Harriss, 1994) and in part
to stratospheric ozone depletion that increased tropospheric OH (Bekki et al.,
1994; Fuglestvedt et al., 1994). Records of changes in the 13C/12C
ratios in atmospheric CH4 during this period suggest the existence
of an anomaly in the sources or sinks involving more than one causal factor
(Lowe et al., 1997; Mak et al., 2000).
There is no consensus on the causes of the long-term decline in the annual
growth rate. Assuming a constant mean atmospheric lifetime of CH4
of 8.9 years as derived by Prinn et al. (1995), Dlugokencky et al. (1998) suggest
that during the period 1984 to 1997 global emissions were essentially constant
and that the decline in annual growth rate was caused by an approach to steady
state between global emissions and atmospheric loss rate. Their estimated average
source strength was about 550 Tg/yr. (Inclusion of a soil sink term of 30 Tg/yr
would decrease the lifetime to 8.6 years and suggest an average source strength
of about 570 Tg/yr.) Francey et al. (1999), using measurements of 13CH4
from Antarctic firn air samples and archived air from Cape Grim, Tasmania, also
concluded that the decreased CH4 growth rate was consistent with
constant OH and constant or very slowly increasing CH4 sources after
1982. However, other analyses of the global methyl chloroform (CH3CCl3)
budget (Krol et al., 1998) and the changing chemistry of the atmosphere (Karlsdottir
and Isaksen, 2000) argue for an increase in globally averaged OH of +0.5%/yr
over the last two decades (see Section 4.2.6 below)
and hence a parallel increase in global CH4 emissions by +0.5%/yr.
The historic record of atmospheric CH4 obtained from ice cores has
been extended to 420,000 years before present (BP) (Petit et al., 1999). As
Figure 4.1e demonstrates, at no time during this
record have atmospheric CH4 mixing ratios approached today�s values.
CH4 varies with climate as does CO2. High values are observed
during interglacial periods, but these maxima barely exceed the immediate pre-industrial
CH4 mixing ratio of 700 ppb. At the same time, ice core measurements
from Greenland and Antarctica indicate that during the Holocene CH4
had a pole-to-pole difference of about 44 ± 7 ppb with higher values
in the Arctic as today, but long before humans influenced atmospheric methane
concentrations (Chappelaz et al., 1997; Figure 4.1d).
Finally, study of CH4 ice-core records at high time resolution reveals
no evidence for rapid, massive CH4 excursions that might be associated
with large-scale decomposition of methane hydrates in sediments (Brook et al.,
2000).
The feedback of CH4 on tropospheric OH and its own lifetime is re-evaluated
with contemporary CTMs as part of OxComp, and results are summarised in Table
4.3. The calculated OH feedback, 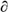 ln(OH)
/ ln(CH4), is
consistent between the models, indicating that tropospheric OH abundances decline
by 0.32% for every 1% increase in CH4. The TAR value for the sensitivity
coefficient s = ln(LT) /
ln(CH4) is then
0.28 and the ratio PT/LT is 1.4. This 40% increase in the integrated effect
of a CH4 perturbation does not appear as a 40% larger amplitude in
the perturbation but rather as a lengthening of the duration of the perturbation
to 12 years. This feedback is difficult to observe, since it would require knowledge
of the increase in CH4 sources plus other factors affecting OH over
the past two decades. Unlike for the global mean tropospheric OH abundance,
there is also no synthetic compound that can calibrate this feedback; but it
is possible that an analysis of the budgets of 13CH4 and
12CH4 separately may lead to an observational constraint
(Manning, 1999). ln(OH)
/ ln(CH4), is
consistent between the models, indicating that tropospheric OH abundances decline
by 0.32% for every 1% increase in CH4. The TAR value for the sensitivity
coefficient s = ln(LT) /
ln(CH4) is then
0.28 and the ratio PT/LT is 1.4. This 40% increase in the integrated effect
of a CH4 perturbation does not appear as a 40% larger amplitude in
the perturbation but rather as a lengthening of the duration of the perturbation
to 12 years. This feedback is difficult to observe, since it would require knowledge
of the increase in CH4 sources plus other factors affecting OH over
the past two decades. Unlike for the global mean tropospheric OH abundance,
there is also no synthetic compound that can calibrate this feedback; but it
is possible that an analysis of the budgets of 13CH4 and
12CH4 separately may lead to an observational constraint
(Manning, 1999).
| Table 4.3: Methane lifetime and feedback on tropospheric
OHa for the 1990s. |
 |
| CTM |
lifetime vs. OH(yr)b
|
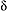 ln(OH)/ln(CH4) ln(OH)/ln(CH4)
|
s=ln(LT)/ln(CH4)
|
PT/LT
|
|
| IASB |
8.1
|
-0.31
|
+0.27
|
1.37
|
| KNMI |
9.8
|
-0.35
|
+0.31
|
1.45
|
| MPIC |
8.5
|
-0.29
|
+0.25
|
1.33
|
| UCI |
9.0
|
-0.34 (-0.38)c
|
+0.30
|
1.43
|
| UIO1 |
6.5
|
-0.33
|
+0.29
|
1.41
|
| UKMO |
8.3
|
-0.31 (-0.34)c
|
+0.27
|
1.37
|
| ULAQ |
13.8
|
-0.29
|
+0.25
|
1.33
|
|
| TAR valued |
9.6
|
-0.32
|
|
1.4
|
|
|