4.2.1.2 Nitrous oxide (N2O)
The globally averaged surface abundance of N2O was 314 ppb in 1998,
corresponding to a global burden of 1510 TgN. N2O abundances are
about 0.8 ppb greater in the Northern Hemisphere than in the Southern Hemisphere,
consistent with about 60% of emissions occurring in the Northern Hemisphere.
Almost no vertical gradient is observed in the troposphere, but N2O
abundances decrease in the stratosphere, for example, falling to about 120 ppb
by 30 km at mid-latitudes.
The known sources of N2O are listed in Table 4.4
with estimates of their emission rates and ranges. As with methane, it remains
difficult to assess global emission rates from individual sources that vary
greatly over small spatial and temporal scales. Total N2O emissions
of 16.4 TgN/yr can be inferred from the N2O global sink strength
(burden/lifetime) plus the rate of increase in the burden. In the SAR the sum
of N2O emissions from specific sources was notably less than that
inferred from the loss rate. The recent estimates of global N2O emissions
from Mosier et al. (1998b) and Kroeze et al. (1999) match the global loss rate
and underline the progress that has been made on quantification of natural and
agricultural sources. The former study calculated new values for N2O
agricultural emissions that include the full impact of agriculture on the global
nitrogen cycle and show that N2O emissions from soils are the largest
term in the budget (Table 4.4). The latter study combined
these with emissions from other anthropogenic and natural sources to calculate
a total emission of 17.7 TgN/yr for 1994.
The enhanced N2O emissions from agricultural and natural ecosystems
are believed to be caused by increasing soil N availability driven by increased
fertilizer use, agricultural nitrogen (N2) fixation, and N deposition;
and this model can explain the increase in atmospheric N2O abundances
over the last 150 years (Nevison and Holland, 1997). Recent discovery of a faster-than-linear
feedback in the emission of N2O and NO from soils in response to
external N inputs is important, given the projected increases of N fertilisation
and deposition increases in tropical countries (Matson et al., 1999). Tropical
ecosystems, currently an important source of N2O (and NO) are often
phosphorus-limited rather than being N-limited like the Northern Hemispheric
terrestrial ecosystems. Nitrogen fertiliser inputs into these phosphorus-limited
ecosystems generate NO and N2O fluxes that are 10 to 100 times greater
than the same fertiliser addition to nearby N-limited ecosystems (Hall and Matson,
1999). In addition to N availability, soil N2O emissions are regulated
by temperature and soil moisture and so are likely to respond to climate changes
(Frolking et al., 1998; Parton et al., 1998). The magnitude of this response
will be affected by feedbacks operating through the biospheric carbon cycle
(Li et al., 1992, 1996).
The industrial sources of N2O include nylon production, nitric acid
production, fossil fuel fired power plants, and vehicular emissions. It was
once thought that emission from automobile catalytic converters were a potential
source of N2O, but extrapolating measurements of N2O emissions
from auto-mobiles in roadway tunnels in Stockholm and Hamburg during 1992 to
the global fleet gives a source of only 0.24 ± 0.14 TgN/yr (Berges et
al., 1993). More recent measurements suggest even smaller global emissions from
automobiles, 0.11 ± 0.04 TgN/yr (Becker et al., 1999; Jiménez
et al., 2000).
The identified sinks for N2O are photodissociation (90%) and reaction
with electronically excited oxygen atoms (O(1D)); they occur in the
stratosphere and lead to an atmospheric lifetime of 120 years (SAR; Volk et
al., 1997; Prinn and Zander, 1999). The small uptake of N2O by soils
is not included in this lifetime, but is rather incorporated into the net emission
of N2O from soils because it is coupled to the overall N-partitioning.
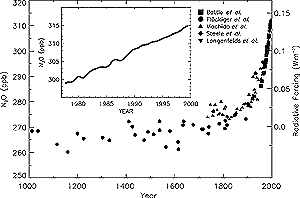
Figure 4.2: Change in N2O abundance for the last 1,000
years as determined from ice cores, firn, and whole air samples. Data sets
are from: Machida et al. (1995); Battle et al. (1996); Langenfelds et al.
(1996); Steele et al. (1996); Flückiger et al. (1999). Radiative forcing,
approximated by a linear scale, is plotted on the right axis. Deseasonalised
global averages are plotted in the inset (Butler et al., 1998b). |
Isotopic (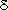 15N
and 18O)
N2O measurements are also used to constrain the N2O budget.
The isotopic composition of tropo-spheric N2O derives from the flux-weighted
isotopic composition of sources corrected for fractionation during destruction
in the stratosphere. Typical observed values are 15N
= 7 ‰ and 18O
= 20.7 ‰ relative to atmospheric N2 and oxygen (O2)
(Kim and Craig, 1990). Most surface sources are depleted in 15N and
18O relative to tropospheric N2O (e.g., Kim and Craig,
1993), and so other processes (sources or sinks) must lead to isotopic enrichment.
Rahn and Wahlen (1997) use stratospheric air samples to show that the tropospheric
isotope signature of N2O can be explained by a return flux of isotopically
enriched N2O from the stratosphere, and no exotic sources of N2O
are needed. Yung and Miller (1997) point out that large isotopic fractionation
can occur in the stratosphere during photolysis due to small differences in
the zero point energies of the different isotopic species, and Rahn et al. (1998)
have verified this latter effect with laboratory measurements. Wingen and Finlayson-Pitts
(1998) failed to find evidence that reaction of CO3 with N2
(McElroy and Jones, 1996) is an atmospheric source of N2O. The use
of isotopes has not yet conclusively identified new sources nor constrained
the N2O budget better than other approaches, but the emerging data
set of isotopic measurements, including measurements of the intra-molecular
position of 15N in N2O isotopomers (Yoshida and Toyoda,
2000) will provide better constraints in the future. 15N
and 18O)
N2O measurements are also used to constrain the N2O budget.
The isotopic composition of tropo-spheric N2O derives from the flux-weighted
isotopic composition of sources corrected for fractionation during destruction
in the stratosphere. Typical observed values are 15N
= 7 ‰ and 18O
= 20.7 ‰ relative to atmospheric N2 and oxygen (O2)
(Kim and Craig, 1990). Most surface sources are depleted in 15N and
18O relative to tropospheric N2O (e.g., Kim and Craig,
1993), and so other processes (sources or sinks) must lead to isotopic enrichment.
Rahn and Wahlen (1997) use stratospheric air samples to show that the tropospheric
isotope signature of N2O can be explained by a return flux of isotopically
enriched N2O from the stratosphere, and no exotic sources of N2O
are needed. Yung and Miller (1997) point out that large isotopic fractionation
can occur in the stratosphere during photolysis due to small differences in
the zero point energies of the different isotopic species, and Rahn et al. (1998)
have verified this latter effect with laboratory measurements. Wingen and Finlayson-Pitts
(1998) failed to find evidence that reaction of CO3 with N2
(McElroy and Jones, 1996) is an atmospheric source of N2O. The use
of isotopes has not yet conclusively identified new sources nor constrained
the N2O budget better than other approaches, but the emerging data
set of isotopic measurements, including measurements of the intra-molecular
position of 15N in N2O isotopomers (Yoshida and Toyoda,
2000) will provide better constraints in the future.
Tropospheric N2O abundances have increased from pre-industrial values
of about 270 ppb (Machida et al., 1995; Battle et al., 1996; Flückiger
et al., 1999) to a globally averaged value of 314 ppb in 1998 (Prinn et al.,
1990, 1998; Elkins et al., 1998) as shown in Figure 4.2.
The pre-industrial source is estimated to be 10.7 TgN/yr, which implies that
current anthropogenic emissions are about 5.7 TgN/yr assuming no change in the
natural emissions over this period. The average rate of increase during the
period 1980 to 1998 determined from surface measurements was +0.8 ± 0.2
ppb/yr (+0.25 ± 0.05 %/yr) and is in reasonable agreement with measurements
of the N2O vertical column density above Jungfraujoch Station, +0.36
± 0.06%/yr between 1984 and 1992 (Zander et al., 1994). Large interannual
variations in this trend are also observed. Thompson et al. (1994) report that
the N2O growth rate decreased from 1 ppb/yr in 1991 to 0.5 ppb/yr
in 1993 and suggest that decreased use of nitrogen-containing fertiliser and
lower temperatures in the Northern Hemisphere may have been in part responsible
for lower biogenic soil emissions. Schauffler and Daniel (1994) suggest that
the N2O trend was affected by stratospheric circulation changes induced
by massive increase in stratospheric aerosols following the eruption of Mt.
Pinatubo. Since 1993, the N2O increase has returned to rates closer
to those observed during the 1980s.
The feedback of N2O on its own lifetime (Prather, 1998) has been
examined for this assessment with additional studies from established 2-D stratospheric
chemical models. All models give similar results, see Table
4.5. The global mean atmospheric lifetime of N2O decreases about
0.5% for every 10% increase in N2O (s = -0.05). This shift is small
but systematic, and it is included in Table 4.1a
as a shorter perturbation lifetime for N2O, 114 years instead of
120 years. For N2O (unlike for CH4) the time to mix the
gas into the middle stratosphere where it is destroyed, about 3 years, causes
a separation between PT (about 114 years) and the e-fold of the long-lived mode
(about 110 years).
| Table 4.4: Estimates of the global nitrous oxide
budget (in TgN/yr) from different sources compared with the values adopted
for this report (TAR). |
 |
| Reference: |
Mosier et al. (1998b)
|
Olivier et al. (1998)
|
SAR
|
TAR
|
| |
Kroeze et al. (1999)
|
|
|
|
|
| Base year: |
1994
|
range
|
1990
|
range
|
1980s
|
1990s
|
|
| Sources |
|
|
|
|
|
|
| Ocean |
3.0
|
1-5
|
3.6
|
2.8-5.7
|
3
|
|
| Atmosphere (NH3 oxidation) |
0.6
|
0.3 -1.2
|
0.6
|
0.3 -1.2
|
|
|
| Tropical soils |
|
|
|
|
|
|
| Wet forest |
3.0
|
2.2 -3.7
|
|
|
3
|
|
| Dry savannas |
1.0
|
0.5 -2.0
|
|
|
1
|
|
| Temperate soils |
|
|
|
|
|
|
| Forests |
1.0
|
0.1 -2.0
|
|
|
1
|
|
| Grasslands |
1.0
|
0.5 -2.0
|
|
|
1
|
|
| All soils |
|
|
6.6
|
3.3 -9.9
|
|
|
| Natural sub-total |
9.6
|
4.6 -15.9
|
10.8
|
6.4 -16.8
|
9
|
|
| Agricultural soils |
4.2
|
0.6 -14.8
|
1.9
|
0.7 -4.3
|
3.5
|
|
| Biomass burning |
0.5
|
0.2 -1.0
|
0.5
|
0.2 -0.8
|
0.5
|
|
| Industrial sources |
1.3
|
0.7 -1.8
|
0.7
|
0.2 -1.1
|
1.3
|
|
| Cattle and feedlots |
2.1
|
0.6 -3.1
|
1.0
|
0.2 -2.0
|
0.4
|
|
| Anthropogenic Sub-total |
8.1
|
2.1 -20.7
|
4.1
|
1.3 -7.7
|
5.7
|
6.9a
|
| Total sources |
17.7
|
6.7 -36.6
|
14.9
|
7.7 -24.5
|
14.7b
|
|
|
| Imbalance (trend) |
3.9
|
3.1 -4.7
|
|
|
3.9
|
3.8
|
|
| Total sinks (stratospheric) |
12.3
|
9 -16
|
|
|
12.3
|
12.6
|
| Implied total source |
16.2
|
|
|
|
16.2
|
16.4
|
|
| Table 4.5: Nitrous oxide lifetime feedback and residence
time. |
|
| Models |
Contributor |
Lifetime |
Sensitivity, |
Decay Time |
| |
|
LT (yr) |
s=ln(LT)/ln(B) |
of mode (yr) |
|
| AER 2D |
Ko and Weisenstein |
111 |
-0.062 |
102 |
| GSFC 2D |
Jackman |
137 |
-0.052 |
127 |
| UCI 1D |
Prather |
119 |
-0.046 |
110 |
| Oslo 2D |
Rognerud |
97 |
-0.061 |
|
|
|