|
2.1.2. Effects of Aircraft Gaseous Emissions
As discussed above, the concentration of ozone is determined by transport from
other locations and by local production and loss. Production and destruction
rates of ozone are strongly influenced by the concentration of the free-radical
catalysts NO and NO2 (NOx)
and OH and HO2 (HOx).
In the presence of NO, ozone is produced as a by-product when CO, CH4,
and other hydrocarbons are oxidized by OH. NOx
also influences the destruction rate of ozone directly as a catalyst (in the
stratosphere) and indirectly as a result of reactions that couple NOx
to other reactive species such as the odd hydrogen radicals, OH and HO2,
and, in the stratosphere, the halogen free radicals chlorine monoxide
(ClO) and bromine monoxide (BrO).
2.1.2.1. Production of Ozone
The production of ozone in the stratosphere is dominated by the photolysis
of oxygen (O2) by sunlight. Because radiation with short wavelengths (less than
242 nm) is screened out by O2 and ozone in the upper atmosphere, this process
is not very important in the troposphere. In the region where commercial aircraft
fly, ozone (O3) is produced mainly from the oxidation of CO:
OH + CO  H + CO2
H + CO2 |
(1) |
| H + O2 + M
HO2 + M |
(2) |
| HO2 + NO
NO2 + OH |
(3) |
| NO2 + sunlight
NO + O |
(4) |
| O + O2 + M
O3 + M |
(5) |
Net: CO + 2 O2
CO2 + O3
(where M represents a gaseous third body such as N2
or O2). The oxidation of CH4
also contributes to ozone formation but is less important in the UT than
reaction 1. |
The rates of these reactions depend directly on the concentrations of NOx and
HOx. Increases in the concentration of NOx from aircraft generally will increase
the rate of ozone production by speeding the oxidation of CO and CH4. Other
aircraft emissions, such as H2O, CO, and NMHCs, are not expected to significantly
affect this background chemistry because natural sources of these compounds
far exceed the perturbation from aviation (Friedl, 1997; Brasseur et al., 1998).
Within the plume, however, the production of particulate and subsequent contrail
and cloud formation may influence NOx and HOx and therefore the production rate
of ozone (see Section 2.1.3).
2.1.2.2. Sources and Sinks of NOx
Aircraft emissions are one of many sources of NOx in the troposphere and the
stratosphere. In the stratosphere, NOx is produced primarily from the oxidation
of nitrous oxide (N2O). N2O is produced by numerous sources, and its concentration
has been increasing at a rate of 0.5-0.8 ppbv yr-1 (IPCC, 1996). As a result
of this source, NOx concentrations in the LS are quite large, increasing from
about 100 pptv at the tropopause to as much as 3000 pptv at an altitude of 20
km. Aircraft exhaust provides an additional source of NOx, but there is no evidence
that this source has appreciably altered the concentration of stratospheric
NOx.
The primary sources of NOx in the troposphere are fossil fuel combustion, biomass
burning, soil emissions, lightning, transport from the stratosphere, ammonia
oxidation, and aircraft exhaust. The largest source is fossil fuel combustion;
95% of its emissions are in the Northern Hemisphere (Lee et al., 1997). Biomass
burning occurs primarily in the continental tropics. Soil emissions come from
microbial denitrification and nitrification processes, the rate depends on soil
type and temperature, ecosystem type, water content, and several other variables
(Matthews, 1983; Müller, 1992; Williams et al., 1992; Yienger and Levy, 1995).
The contribution of NOx produced by lightning is very uncertain because it is
extremely difficult to measure directly. The distribution around thunderstorms
is highly variable because NO production differs for cloud-to-cloud and cloud-to-ground
strikes. Oxidation of ammonia (NH3) of NO occurs at the surface, mainly in the
tropics. Although the sources of NH3 are fairly well known, the rates of reactions
that result in NO are uncertain (Lee et al., 1997), and, under some conditions,
ammonia oxidation can be a sink for NOx.
Various studies have estimated the emission of NOx from these sources (Müller,
1992; Atherton et al., 1996; Lee et al., 1997). Additionally, estimates for
individual sources have been produced: fossil fuel combustion (Dignon, 1992;
Benkovitz et al., 1996); biomass burning (Crutzen and Andreae, 1990; Müller,
1992; Atherton, 1995); soil emissions (Matthews, 1983; Williams et al., 1992;
Yienger and Levy, 1995); lightning (Price and Rind, 1992; Levy et al., 1996;
Ridley et al., 1996); aircraft exhaust (Wuebbles et al., 1993; Metwally, 1995;
Baughcum et al., 1996; Gardner et al., 1997); and transport from the stratosphere
(Kasibhatla et al., 1991; Wang et al., 1998a).
Compilations and evaluations of emission rates (Friedl, 1997; Lee et al., 1997)
have emphasized that the contribution from lightning remains highly uncertain.
The relative impact of NOx produced from aviation is critically dependent on
the strength of this source. Despite this uncertainty, it is likely that averaged
over the globe, the lightning source of NOx in the UT is 2 to 8 times as large
as the aircraft source. Although lightning, aircraft emissions, and transport
of NOx from the boundary layer during deep convection clearly provide significant
sources of NOx to the UT, the contribution of each in determining the total
NOx concentration is not well understood.
In situ observations have been made in the UT to help identify and quantify
sources of NOx. The concentration of NO measured
in these field campaigns is extremely variable. Based on a number of in situ
aircraft missions, however, typical NO concentrations in the UT are in the range
50-200 pptv (Emmons et al., 1997; Schlager et al., 1997; Tremmel et al., 1998).
Because of the large variability in the atmosphere, these relatively sparse
observations cannot quantitatively define the strengths of the various NOx
sources in the UT. Recent aircraft campaigns-for example, SONEX, POLINAT-2,
NOXAR-should, however, provide important constraints
on the role of aircraft in perturbing NOx. For
example, a full year of observations of NO in the UT and lowermost stratosphere
obtained onboard a commercial airplane have demonstrated the importance of NOx
sources associated with convection (Brunner et al., 1998).
The NOx species belong to a family of chemicals collectively called reactive
nitrogen (NOy). This family includes species that are coupled to NOx on time
scales of one day to several weeks, such as HNO4, nitrogen pentoxide (N2O5),
nitric acid (HNO3), and, in the troposphere, peroxyacetylnitrate (PAN). The
primary form of reactive nitrogen emitted into the atmosphere is NO. Once NO
is in the atmosphere, it undergoes rapid interconversion with NO2 and then slower
conversion to other reactive nitrogen species such as HNO3.
|
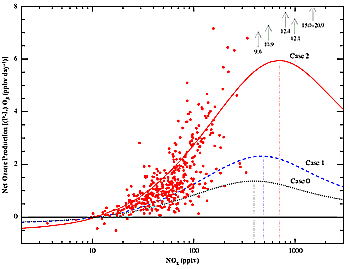
Figure 2-1: Net ozone production (24-hr average) as a function of NOx in the upper troposphere
(adapted from Jaeglé et al., 1998).
|
Reactions occurring on aerosol surfaces also play an important role in the
chemical partitioning of NOy. It is well-established that NOx concentrations
are reduced and HNO3 concentrations are enhanced as a result of heterogeneous
chemistry on aerosol surfaces. It is also possible that reactions occurring
on soot may play the opposite role, converting HNO3 back to NOx (Section
2.1.3).
Observations of NOy species have demonstrated that our understanding of the
sources of NOy and the partitioning of NOy in the stratosphere is relatively
good (within ± 30%) (Rinsland et al., 1996; Gao et al., 1997). Photochemical
models of the partitioning and distribution of NOy in the UT, however, have
typically demonstrated poor agreement with observations. Because the reactions
that convert NOx, PAN, and HNO3 occur on time scales similar to transport, it
is not clear whether the observed differences are caused by errors in the chemical
description or by dynamic influences that are not captured in the models (Prather
and Jacob, 1997).
2.1.2.3. Sources and Sinks of OH and HO2
HOx is produced in the UT and LS via a number of processes. One important primary
source of OH is the reaction of water vapor with O(1D) produced in the photolysis
of ozone:
| O3 + sunlight
O(1D) + O2 |
(6a) |
| O(1D) + H2O
OH + OH |
(7) |
Additional sources of OH and HO2 in the troposphere include the photolysis
of acetone (Singh et al., 1995), peroxides (Chatfield and Crutzen, 1984), and
formaldehyde. In the stratosphere, the photolysis of HOBr and HNO3 produced
in reactions occurring on aerosols may also be important HOx sources (Hanson
et al., 1996). The existence of important primary HOx sources in addition to
reactions 6a and 7 has been demonstrated by recent measurements of OH and HO2
in the UT and LS (McKeen et al., 1997; Jaeglé et al., 1997, 1998; Brune et al.,
1998; Wennberg et al., 1998). These primary sources of OH can be further amplified
during the oxidation of CH4 and other hydrocarbons.
OH is chemically coupled to HO2 by reactions such as 1-3 that convert these
species on time scales of seconds to minutes. Removal of HOx (leading to the
formation of H2O) occurs on a longer time scale (5-30 min) and is dominated
by processes such as:
| OH + HO2
H2O + O2 |
(8) |
| |
|
| OH + NO2 + M
HNO3 + M |
(9) |
| OH + HNO3
H2O + NO3 |
(10) |
| Net: OH + OH + NO2
H2O + NO3
|
|
| |
|
| HO2 + NO2
+ M HNO4
+M |
(10) |
| OH + HNO4
H2O + NO2
+ O2 |
(12) |
| Net: OH + HO2
H2O + O2 |
|
| |
|
| HO2 + HO2
+ M H2O2
+ O2 + M |
(13) |
| OH + H2O2
H2O
+ HO2 |
(14) |
| Net: OH + HO2
H2O + O2 |
|
NOx and HOx are therefore linked by a number of important reactions (3,9,10-12);
the concentration of each depends on the concentration of the other.
2.1.2.4. The Response of Ozone to NOx in the Upper Troposphere
Figure 2-1 illustrates how the background photochemistry
works to change ozone in the UT in response to variation in the concentration
of NOx (Jaeglé et al., 1998). The points are calculated from in situ measurements
of HOx and NO obtained during the SUCCESS campaign. The lines show how the calculated
response varies with assumptions about HOx source strength. The response includes
ozone production via reactions 4 and 5 and ozone destruction primarily via:
| O3 + HO2
OH + 2 O2 |
(15) |
In all models, the net ozone production rate (production rate - loss rate)
increases rapidly with NOx until a maximum is reached. At NOx concentrations
larger than 500 pptv, the net rate of ozone production is expected to decrease
with increasing NOx. Depending on the background concentration of NOx, additions
of NOx from aviation can increase or decrease the net ozone production rate.
Thus, the background concentration of NOx determines both the magnitude and
the sign of the perturbation. Field measurements of NOx in the middle and upper
troposphere typically have found NOx to be 50-200 pptv (Emmons et al., 1997;
Schlager et al., 1997; Tremmel et al., 1998). At these concentrations, the rate
of net ozone production increases almost linearly with NOx.
Figure 2-1 also illustrates that the rate of ozone
production depends critically on HOx source strength. In this figure, the calculation
in Case 0 assumes that the primary source of HOx is limited to the reaction
of O1D with H2O and CH4. This assumption is made in many (but not all) of the
chemical transport models used to assess the effect of aviation in Section
2.2.1 and Chapter 4. Case 1 includes an additional source of HOx from acetone
photolysis (Singh et al., 1995), assuming acetone is present at a concentration
roughly consistent with recent measurements in the UT (Singh et al., 1995; Arnold
et al., 1997). Case 2 assumes that in addition to acetone, peroxides and formaldehyde
are transported to the UT by convection (Chatfield and Crutzen, 1984; Jaeglé
et al., 1997). It is clear from observations of HOx obtained from the DC-8 (Brune
et al., 1998) and the ER-2 (Wennberg et al., 1998) that HOx sources in addition
to O1D + H2O are needed to explain measured concentrations of OH and HO2. The
effect of these HOx sources is most pronounced in UT air when the water vapor
mixing ratio is less than 100 ppmv. At median NOx concentrations observed during
these campaigns-50-100 pptv, typical of the UT (Brunner et al., 1998)-the net
ozone production rate is calculated to be 1-2 ppbv per day. This rate is significantly
faster than would be calculated assuming only the simple O1D HOx chemistry.
The sensitivity of the net ozone production rate to assumptions about the sources
of odd hydrogen is high and remains an area of significant uncertainty. The
budget of acetone, for example, is poorly understood, and relatively few measurements
of its concentration have been made in the UT. Observational constraints on
the HOx chemistry of the UT are just now becoming available; the number of measurements
is expected to increase greatly over the next few years.
2.1.2.5. The Response of Stratospheric Ozone Destruction
to NOx
The concentration of NOx also influences the rate at which ozone is destroyed
in the atmosphere, particularly in the stratosphere and in the lower troposphere.
In the stratosphere, because of the abundance of ozone and lower pressures,
the concentration of atomic oxygen is sufficiently large that NOx can catalytically
destroy ozone:
| O3 + sunlight
O + O2 |
(6b) |
| O + NO2
NO + O2 |
(16) |
| NO + O3
NO2 + O2 |
(17) |
| Net: 2 O3
3 O2 |
|
Reactions of the HOx family also destroy ozone. In particular, reaction 15
leads to significant ozone loss in the LS and in the troposphere. Reaction 3
is in competition with reaction 15, so the rate of ozone loss by HOx decreases
with increasing NO.
|
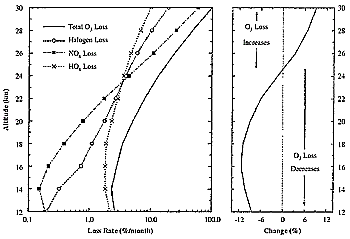
Figure 2-2: A calculation of the rate of ozone loss in the lower stratosphere for springtime
mid-latitude conditions during March.
|
Finally, ozone loss by halogen chemistry is important in the stratosphere.
During winter, particularly in polar regions, it can dominate all other chemical
destruction mechanisms. As with HOx chemistry, NOx interferes with this chemistry
by binding to the reactive chlorine radical ClO:
| ClO + NO2 + M
ClONO2 + M. |
(18) |
As a result of these coupling reactions, changes in the concentration of NOx
can lead to increased or decreased rates of stratospheric ozone destruction.
When NOx is low-as it is in most of the LS during winter, fall, and spring-most
of the ozone loss occurs through HOx and halogen chemistry. Under these conditions,
enhancements of NOx will decrease ozone destruction. On the other hand, at higher
altitudes and during summer, NOx-catalyzed ozone loss (reactions 16-17) can
dominate the removal of lower stratospheric ozone, so enhancements in NOx will
speed ozone loss (Brühl et al., 1998). These effects have been demonstrated
by direct measurements of free radicals in the stratosphere (Wennberg et al.,
1994; Jucks et al., 1997).
This chemistry is illustrated in Figure 2-2. A
calculation is shown for typical mid-latitude springtime conditions. This entire
profile is within the stratosphere, where catalytic ozone loss competes with
and can exceed photochemical production. The left panel shows the fraction of
ozone destroyed during the month of March as a result of catalysis by NOx
(squares), halogens (circles), and HOx (crosses).
For this latitude and season, the loss is dominated by halogen and hydrogen
oxides below 20 km, whereas above 25 km, nitrogen oxides are most important.
To illustrate how changes in NOx perturb this
chemistry, the right panel shows the effect of a uniform 20% increase in the
concentration of NOx. In regions where NOx
is high, ozone destruction increases. On the other hand, the opposite occurs
in the LS because the increased NOx decreases
the loss of ozone by hydrogen and halogen radicals. Thus, as with the production
rate of ozone in the troposphere, the response of ozone destruction with changes
in NOx is highly nonlinear. Because the photochemical
lifetime of ozone in the LS is very long, the concentration of ozone in this
region of the atmosphere is strongly influenced by transport. The change in
ozone loss rates illustrated in Figure 2-2 does
not translate directly into a change in ozone. For example, for a uniform 20%
increase in NOx, enhanced loss rates at high
altitudes will reduce the transport of ozone to the LS. As a result, ozone concentrations
in the LS can decrease even when the local ozone loss rate slows. Thus, the
change in the ozone column with added NOx is
very sensitive to the altitude distribution of the perturbation. The subsonic
aircraft fleet adds NOx only to the lowermost
stratosphere (< 13 km), where large-scale dynamics tend to prevent advection
to higher altitude. As a result, injection of NOx
by the present fleet is thought to increase ozone in the LS.
2.1.2.6. Net Effects on Ozone
If the major direct impact of aircraft on the chemistry of the UT and lowermost
stratosphere (below approximately 16 km) is an increase in the concentration
of NOx, we can say with high confidence that the ozone concentrations in this
region will be higher than they would be in the absence of aviation. This increase
occurs because NO speeds the catalytic oxidation rate of CO and reduces the
destruction rate of ozone by HOx and halogens (primarily in the stratosphere).
In this context, it is important to note that the conventional troposphere-stratosphere
boundary (i.e., the tropopause), reflecting important changes in atmospheric
dynamics, does not coincide with the separation between net positive and negative
NOx-induced changes in ozone (see right panel of Figure
2-2). This distinction is important when considering future aviation scenarios
that include a significant supersonic component.
The actual quantitative change in ozone from present aircraft operation is
very sensitive to the meteorology of this region of the atmosphere; longer residence
times will lead to larger NOx increases and therefore higher ozone. As a result,
only coupled chemical and dynamic models can estimate how large an increase
is expected. To accurately predict the perturbation, these models must accurately
describe the background NOx concentration, the magnitude of the aircraft-induced
NOx perturbation, the sources of HOx in this region of the atmosphere, and the
meteorology. Finally, because ozone itself is relatively long-lived in the UT
and LS, its concentration is strongly influenced by transport (as discussed
in Section 2.1.1.3.3). The transport of ozone between
different regions of the atmosphere significantly confounds attempts to assign
causality to local ozone trends. As described in Section 2.1.3,
the production of particulates and aircraft-induced formation of clouds may
be important for ozone. Chemical processes occurring in aerosols and on ice
clouds may lead to increases in chlorine radical abundance in the stratosphere
and suppress NOx throughout the region.
|