|
4.2.4 Tropospheric O3
Tropospheric O3 is a direct greenhouse gas. The past increase in
tropospheric O3 is estimated to provide the third largest increase
in direct radiative forcing since the pre-industrial era. In addition, through
its chemical impact on OH, it modifies the lifetimes of other greenhouse gases,
such as CH4. Its budget, however, is much more difficult to derive
than that of a long-lived gas for several reasons. Ozone abundances in the troposphere
typically vary from less than 10 ppb over remote tropical oceans up to about
100 ppb in the upper troposphere, and often exceed 100 ppb downwind of polluted
metropolitan regions. This variability, reflecting its rapid chemical turnover,
makes it impossible to determine the tropospheric burden from the available
surface sites, and we must rely on infrequent and sparsely sited profiles from
ozone sondes (e.g., Logan, 1999). The total column of ozone is measured from
satellites, and these observations have been used to infer the tropospheric
ozone column after removing the much larger stratospheric column (e.g., Fishman
and Brackett, 1997; Hudson and Thompson, 1998; Ziemke et al., 1998). The current
burden of tropospheric O3 is about 370 Tg(O3), which is
equivalent to a globally averaged column density of 34 DU (Dobson Units, 1 DU
= 2.687 1016 molecules/cm-2)
or a mean abundance of about 50 ppb, see Table 4.9. 1016 molecules/cm-2)
or a mean abundance of about 50 ppb, see Table 4.9.
| Table 4.9: Estimates of the change in tropospheric
ozone since the pre-industrial era from various sources compared with the
values recommended in this report. |
 |
| Current climatology of tropospheric ozone (Park et
al., 1999): |
| |
Global mean tropospheric O3 : 34 DU = 370 Tg(O3)
content in the Northern Hemisphere = 36 DU, in the Southern Hemisphere =
32 DU. |
|
| SAR recommendation: |
| |
“50% of current Northern Hemisphere is anthropogenic” gives
pre-industrial global mean content = 25 DU. |
| Increase = +9 DU |
|
| 19th & early 20th century observations:a |
| |
Assume Northern Hemisphere tropospheric ozone has increased
uniformly by >30 ppb. |
| Increase = +10 to +13 DU |
|
| Survey of CTM simulated change from pre-industrial:b |
| |
DU increase |
Model |
Reference |
| |
9.6 |
UIO |
Berntsen et al. (1999) |
| |
7.9 |
GFDL |
Haywood et al. (1998) |
| |
8.9 |
MOZART-1 |
Hauglustaine et al. (1998) |
| |
8.4 |
NCAR/2D |
Kiehl et al. (1999) |
| |
9.5 |
GFDL-scaled |
Levy et al. (1997) |
| |
12.0 |
Harvard/GISS |
Mickley et al. (1999) |
| |
7.2 |
ECHAM4 |
Roelofs et al. (1997) |
| |
8.7 |
UKMO |
Stevenson et al. (2000) |
| |
8.0 |
MOGUNTIA |
VanDorland et al. (1997) |
| Increase = +7 to +12 DU (model range) |
|
| TAR recommendation: |
| |
Pre-industrial era global mean tropospheric O 3 has increased
from 25 DU to 34 DU. This increase, +9 DU, has a 67% likely range of 6 to
13 DU. |
| Increase = +9 DU (+6 to +13 DU) |
|
The sources and sinks of tropospheric ozone are even more difficult to quantify
than the burden. Influx of stratospheric air is a source of about 475 Tg(O3)/yr
based on observed correlations with other gases (Murphy and Fahey, 1994; McLinden
et al., 2000). The in situ photochemical sources are predicted to be many times
larger, but are nearly balanced by equally large in situ chemical sinks (see
discussion on CTM modelling of tropo-spheric O3 in Sections
4.4 and 4.5, Table 4.12).
Photochemical production of ozone is tied to the abundance of pollutants and
thus varies widely over a range of spatial scales, the most important of which
(e.g., biomass burning plumes, urban plumes, aircraft corridors, and convective
outflows) are not well represented in most global CTMs and cannot be quantified
globally with regional models. The dominant photochemical sinks for tropospheric
O3 are the catalytic destruction cycle involving the HO2
+ O3 reaction and photolytic destruction by pathways involving the
reaction of O(1D), a product of O3 photodissociation.
The other large sink, comparable in magnitude to the stratospheric source, is
surface loss mainly to vegetation. Another loss of O3 is observed
under certain conditions in the polar marine boundary layer, notably at the
end of Arctic winter. It indicates reactions involving halogen radicals and
aerosols (Oum et al., 1998; Dickerson et al., 1999; Impey et al., 1999; Platt
and Moortgat, 1999; Prados et al., 1999; Vogt et al., 1999). The contribution
of these processes to the global budget is not yet quantified, but is probably
small.
Atmospheric measurement campaigns, both at surface sites and with aircraft,
have focused on simultaneous observations of the many chemical species involved
in tropospheric O3 production. Primary areas of O3 production
are the mid-latitude industrialised and tropical biomass burning regions. For
example, the North Atlantic Regional Experiment (NARE) and the Atmosphere Ocean
Chemistry Experiment (AEROCE) showed that the prevailing westerly winds typically
carry large quantities of ozone and precursors from the eastern USA over the
North Atlantic, reaching Bermuda and beyond (e.g., Dickerson et al., 1995; Penkett
et al., 1998; Prados et al., 1999). The Pacific Exploratory Missions (PEM: Hoell
et al., 1997, 1999) measured extensive plumes of pollution including ozone and
its precursors downwind of eastern Asia. Convective transport of emissions from
biomass burning affect the abundance of O3 in the mid- and upper
troposphere (Pickering et al., 1996). Emissions by tropical fires in South America
and southern Africa have been identified as the cause of enhanced O3
over the South Atlantic (Thompson et al., 1996), and the effects of biomass
burning were seen in the remote South Pacific in PEM Tropics A (Schultz et al.,
1999; Talbot et al ., 1999). Due to the widely varying chemical environments,
these extensive studies provide a statistical sampling of conditions along with
a critical test of the photochemistry in CTM simulations, but they do not provide
an integrated budget for tropospheric O3. An example of such model-and-measurements
study is given in the Section 4.2.6 discussion of
tropospheric OH.
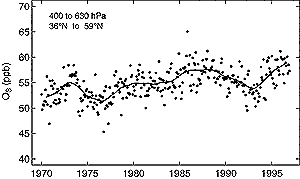
Figure 4.8: Mid-tropospheric O3 abundance (ppb) in
northern mid-latitudes (36°N-59°N) for the years 1970 to 1996. Observations
between 630 and 400 hPa are averaged from nine ozone sonde stations (four
in North America, three in Europe, two in Japan), following the data analysis
of Logan et al. (1999). Values are derived from the residuals of the trend
fit with the trend added back to allow for discontinuities in the instruments.
Monthly data (points) are shown with a smoothed 12-month-running mean (line).
|
Recent trends in global tropospheric O3 are
extremely difficult to infer from the available measurements, while trends in
the stratosphere are readily identified (Randel et al., 1999; WMO, 1999). With
photochemistry producing local lifetimes as short as a few days in the boundary
layer, the local measurement of tropo-spheric O3 does not reflect
the abundance over the surrounding continent, and a surface measurement is not
representative of the bulk of the troposphere above. Thus it is not contradictory
for decadal trends in different atmospheric regions to be different, e.g., driven
by the regional changes in pollutants, particularly NOx. Ozone sondes
offer the best record of O3 throughout the troposphere, although
measurements at many stations are made only weekly (infrequently for a variable
gas like O3). Weekly continuous data since 1970 are available from
only nine stations in the latitude range 36°N to 59°N (Logan et al.,
1999; WMO, 1999). Different trends are seen at different locations for different
periods. Most stations show an increase from 1970 to 1980, but no clear trend
from 1980 to 1996. A composite record of the mid-tropospheric O3
abundance from 1970 to 1996 from the nine stations is taken from the analysis
of Logan et al. (1999) and presented in Figure 4.8.
There is no obvious linear increase in O3 abundance over this period,
although the second half of this record (about 57 ppb) is clearly greater than
the first half (about 53 ppb). Of the fourteen stations with records since 1980,
only two, one in Japan and one in Europe, had statistically significant increases
in mid-tropospheric O3 between 1980 and 1995. By contrast, the four
Canadian stations, all at high latitudes, had significant decreases for the
same time period. Surface O3 measurements from seventeen background
stations also show no clear trend, even in the northern mid-latitudes (Oltmans
et al., 1998; WMO, 1999). The largest negative trend in surface O3
was -0.7 ± 0.2%/yr at the South Pole (1975 to 1997), while the largest
positive trend was +1.5 ± 0.5%/yr at Zugspitze, Germany (1978 to 1995).
This ambiguous record of change over the past two decades may possibly be reconciled
with the model predictions (see Section 4.4) of increasing
tropospheric O3 driven regionally by increasing emissions of pollutants:
the growth in NOx emissions is expected to have shifted from North
America and Europe to Asia.
The change in tropospheric O3 since the pre-industrial era is even
more difficult to evaluate on the basis of measurements alone. Since O3
is reactive, atmospheric abundances cannot be retrieved from ice cores. Recent
evaluations of surface measurements in the 19th and early 20th century in Europe
(Volz and Kley, 1988; Staehelin et al., 1994, 1998; Harris et al., 1997) indicate
much lower O3 abundances than today, yet the scaling of these data
to a tropospheric O3 burden, even for northern mid-latitudes, is
not obvious. In the SAR, these data were used to make a rough estimate that
O3 abundances in the Northern Hemispheric troposphere have doubled
since the pre-industrial era. A similar difference, of 10 to 13 DU when globally
averaged, is obtained using the climatology given by Park et al. (1999) for
tropospheric O3 today and a parallel one with abundances adjusted
to match the 19th century measurements in the Northern Hemisphere. CTM calculations
predict that current anthropogenic emissions of NOx, CO, and VOC,
as well as the increase in CH4 should have increased tropospheric
O3 by a similar amount, primarily in the Northern Hemisphere. A recent
survey of CTM studies gives global average increases ranging from 8 to 12 DU,
although this small range does not adequately represent the uncertainty. These
results are summarised in Table 4.9. Based on measurements,
analyses, and models, the most likely increase in tropospheric O3
was 9 DU globally averaged, with a 67% confidence range of 6 to 13 DU. For some
of the emissions scenarios considered here, tropospheric O3 is expected
to increase even more in the 21st century as emissions of its precursors - NOx,
CO and VOC - continue to grow (see Section 4.4).
|